shelxtl软件是一款功能强大的解晶体软件,软件包含XPREP,XS,XP,XL,XCIF五大程序,采用精确地分析计算技术,可以帮助用户快速分析晶体的结构和详细数据,想要更好的分析晶体的结构和详细数据。在SHELXTL结构分析过程中,主要涉及到三个数据文件:name.hkl;name.ins;name.res,其中INS和RES文件具有相似的数据格式,区别只是INS是指令(instruction)文件,它主要是充当XS及XL的输入文件,而RES是结果(result)文件,主要保存XS及XL的结果,RES中包含有直接法XS或最小二乘法XL产生的差Fourier峰。HKL文件是ASCII型的衍射点数据文件,包含H,K,L,I和o(I),其中HKL文件的格式决定了它不能进行除了DIFABS型校正之外任何校正,INS和RES文件中主要包含单胞参数,分子式(原子类型),原子位置坐标及XL指令等,它们是由一些指令定义的ASCII文件。
1、数据校正-SADABS
SMART CCD由于设备的特殊性使得它不具有四圆一样的PSI校正(由于晶体外观的不对称性),但由于在CCD所收集的数据中有很多对的等效点,因而也可拟合出一条经验校正曲线。SADABS就是Sheldrick特别为CCD数据编写的校正程序。
由于SADABS使用等效点,因而要求输入正确的Laue群,只有正确的Laue群才能保证校正的正确性。
SADABS还提供了与θ有关的校正——所谓的球形校正,它的原理就是在不同的θ角衍射时,X射线通过的光程不同,因而吸收也不同,该校正要求μR值,其中R为晶体几何尺寸中最小的边长,而晶体的吸收因子μ是由化合物的分子式决定的,因而只有在结构完全解释出来之后才能进行这个校正,而这种校正目前有些刊物是要求提供的。也正因为如此,要求保留最原始的数据文件*.RAW。
2、数据处理-XPREP
XPREP主要完成数据处理工作,它要求存在RAW或HKL文件,它的使用命令是:
Xprep name
它是一个交互式菜单驱动程序并提供了一个缺省运行过程:
1.从name.hkl文件(若存在)或name.raw文件中读入衍射点
2.从name.p4p或键盘获得单胞参数及误差;
3.判断晶格类型
3、寻找最高对称性
单胞参数只是晶体对称性的外在表现形式,衍射点的对称才是晶体对称性的内在表现。虽然SMART中也对晶格类型进行判断,但由于CCD中搜寻衍射点的对称性的代价较为昂贵,通常在收集数据时不检测衍射点的对称性,这样导致在收集数据时所判断的对称性不准确,而且由于此时进行指标化的衍射点未必很好,导致某些轴之间的偏差比设定的偏差大从而不能得到真正的对称性,即使选定的对称性比实际的对称性要低。而在SAINT以大量的衍射点精修单胞参数之后,单胞参数趋向真实值,此时再对单胞参数进行转化,可以更准确地得到晶体的对称性。
4、确定空间群
XPREP按照选定的晶系,晶格类型,E值统计,消光特点来判断空间群,并给出了可能的空间群及其对应的综合因子CFOM,CFOM越小,空间群的可能性越大,一般CFOM小于1表明建议的空间群很大可能是正确的,而CFOM大于10则表明可能是错误的,通常CFOM小于10的空间群是可以接受的
5、输入分子式
SHELXTL在进行结构解释时,分子式并不十分重要,重要的只是原子的种类,当然在有有机基团存在的情况下只提供原子种类是难以进行结构解释的,必须提供有机基团的结构类型,最简单的例子:六元环可能是苯环,也可能是吡啶环甚至其它的环,若不知基团类型是无法解释的。
但在产生结构报表时还是需要准确的分子时,甚至在进行与θ有关的校正时也需要准确的分子式。
在输入原子种类之后,XPREP将产生name.ins及name.hkl文件,到此完成数据处理工作。
6、结构解释——XS
除非有异质同晶的化合物结构(可以套用它的结构),否则任何化合物的结构解释都必须对结构进行初解释。SHELXTL中XS可通过①直接法;②Patterson法;③碎片法对结构进行解释,它的运行命令如下:
xs name
它要求存在name.ins及name.hkl两个文件,并将产生name.res文件,在name.res文件中,XS自动按照所给的原子种类把最强的峰命名为最重的原子,并把后续的峰按照其强度进行可能的命名,同时还进行结构修正,产生更多的差Fourier峰。在某些情况下XS结果是极其准确的,它可以直接得到大部分结构(直接法),而这些结构在后续的差Fourier峰中都未必看的更清楚。
表征直接法质量的参数有:CFOM及RE,它们越小表明直接法越成功,通常情况下CFOM在0.1,RE在0.3以下表明直接法可能是成功的。
但直接法也有其局限性,特别是对于那些单斜晶系有心空间群,此时可把空间群降低成无心结构但要记住最后必须把它还原成有心结构,或者可使用Patterson法。在有超过Na的重原子存在的条件下,Patterson法可以给出较好的结果,但Patterson不进行结构修正,也没有很好的表征参数。
32位安装教程
1.双击打开“Disk1”文件夹,并点击“setup.exe”
2.进入正式的安装,可直接跳过内容浏览,直接点击“Next”
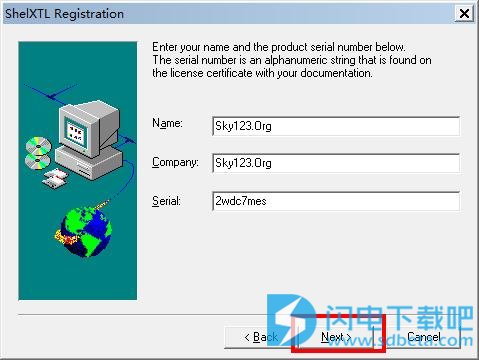
3.进行信息的相关输入,具体信息的输入:
1)NAme和Company尽量不要修改,建议默认
2)seial(序列号):2wdc7mes
4.进行安装路径的选择,默认路径为C:\SAXI,若用户需要更改路径,可点击“Browse”更换路径,路径更为完毕可点击“Next”
5.之后直接点击下一步下一步知道安装完成即可
64位安装教程
若你的系统为win8 64位或win7 64位,那么直接装shelxtl是不可以使用的,需要改一下电脑的环境变量。
打开计算机,右键属性,单击高级系统设置,点击环境变量,单击系统变量下的新建,分别在变量名和值中输入一下信息。
1.变量名path 值%SystemRoot%\system32;%SystemRoot%;%SystemRoot%\System32\Wbem;C:\SAXI\SXTL;;C:\SAXI;C:\Program Files(×86)\Common Files\TTKN\Bin(注意首先找到C盘下的Program Files(×86)这个文件夹,有的在 Files(×86)中间有空格,那么也要把空格输入,就是保证与自己电脑里的文件夹一致,如果不一致则无法进行晶体解析)
2.变量名SAXI$ROOT:值C:\SAXI\
3.变量名SAXI$SITE:值 win8 (此处随意输入)
4.变量名SAXI$USERNAME:值user(此处随意输入)
5.变量名SXTL:值C:\SAXI\SXTL\SXTL.INI
6.变量名SXTL$SYSTEM:值C:\SAXI\SXTL\
之后确定,即可
(注意冒号不要丢掉哦)
一、解单晶的基本程序
1.XPREP*输入分子式
2.XS*
3.XL*(结构修正)
1)refine之前的工作
EDIT *.RES
改L.S.4-10FMAP 2 ( )60 PLAN 20
方法1
打开 *.RES,把温度因子高的点删去
方法二 XP
READ*
FMOL
PROJ
PICK 杀点
FILE *
QUIT
4.XL *
(5,6 可循环多次, 直到修出理想结构图)
5.XP
READ *
FMOL
PROJ
PICK 命名/ NAME
FILE *
QUIT
6.EXIT
二、用WINDOWS 系统的PLATON软件找正确或标准的空间群
1.打开FILE
2.进入PLATON\ SHORTCUT 双击CAL-ALL
3.关闭PLATON
4.假DOS下进入LIS及RES文件, 看空间群是否改过
5.EXIT
6.打开PLATON\PLATON MENU\ADDSYM–SHX , 双击
7.进入DOS系统
三、ANIS
1.EDIT *.RES
改ZERR
SFAC
UNIT
加L.S. 10
FMAP 2 ( ) 60
PLAN 20
改原子顺序号
2.XP
READ*
FMOL
PROJ
PICK
FILE*
QUIT
3.XL*
4.EDIT*.RES下加ANIS
5.XL*
6.XP
READ*
FMOL
PROJ
HADD
FILE*
QUIT
7.XL*
8.EDIT*.RES下换WEIGHT至不变
四、CIF及TEX的产生
1.EDIT *.RES下加ACTA
2.XL *3.XCIF4.输入文件名*
5.STRUCTURE CODE [?]: 选T
6.*.TEX
7.多次回车
8.FILE NAME EXTENSION FOR XCIF: DEF
9.Q
10.EDIT
*.TEX
一、选择原子或(Q)峰的方法
1.光标指在欲选择位置,点S键,可依次选择所有的要选择的原子或Q峰
2.光标指在欲选择位置,右手键点“Select”,可依次选择原子
3.用Select菜单(或背景右手键点“Select”)可选择部分和所有的原子或Q峰,顺序与原顺序相同
4.按住“Shift”键,左手键拖出一个选择框并同时轻开,可选择一个或多个原子
5.点refine菜单,系统会提示有Q峰没命名,并选择所有Q峰,顺序与原来顺序相同
二、删除原子或(Q)峰的方法
1.光标指在欲删除位置(原子或键),点K键
2.光标指在欲删除位置,右手键点Bonds and Angles,再在对话框中点“Delete”
3.光标指在欲删除位置,右手键点“Delete”
4.先选择,再点Select菜单中的Kill selected e在ins文件删除
三、恢复删错的原子或(Q)峰的方法
点“Edit>Restore Killed Atom”,先选择要恢复的原子(或Q峰),再点“Restore”
四、命名(标记)原子或(Q)峰的方法
1.先选择,再用Lablels菜单中的Group label
2.光标指在要命名的位置,用右手键的Edit
3.光标指在欲命名位置,右手键点Bonds and Angles,再在对话框中点“Rename”
单晶衍射实验室为用户提供了下列数据文件:
1、*1.raw,*2.raw… CCD最原始文件,为校正而保留
2、*._ls 记录数据处理文件,包含数据完成度及最后精修单胞参数所用的衍射点
3、*.abs 校正结果文件,主要包含Tmin,Tmax
4、*.hkl 经SADABS校正后的衍射点文件
5、*.p4p 矩阵文件,包含单胞参数
(shelxtl 2014无法运行解决办法)
方法一
project→delete掉所有project,hkl文件不行,试试p4p文件。:hand::hand::hand::hand:
方法二
Project name换一个,有重名的










