Shelxtl破解版是一款功能强大的解晶体软件。Shelxtl软件包含XPREP,XS,XP,XL,XCIF五大程序,使用可帮助用户快速对数据进行更改、合并和读入等操作处理,轻松进行对称性的快速寻找,对Patterson界面显示进行计算的操作。帮助用户对要或者已知的空间群进行确定输入 ,也可对晶格类型进行快速的重心设置以及计算的指令文件进行建立。Shelxtl采用精确地分析计算技术,可以帮助用户快速分析晶体的结构和详细数据。本次我们带来的是最新破解版,含详细的安装破解教程!
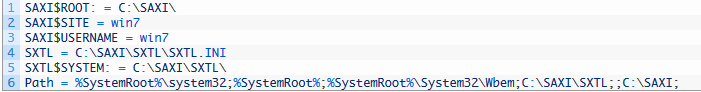
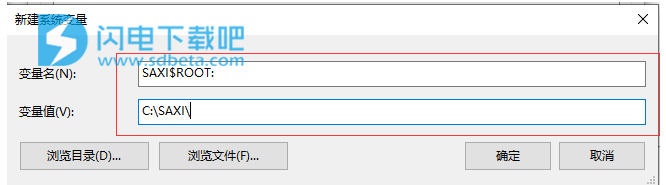
Path=%SystemRoot%\system32;%SystemRoot%;%SystemRoot%\System32\Wbem;C:\SAXI\SXTL;;C:\SAXI;
5、完成啦,试试运行一下~
内容介绍
SHELXT - 新的小分子(SM)结构解决方案。
SHELXS - SM结构解决方案的经典直接方法。
SHELXL - SM和MM的改进,或多或少与SHELX76和SHELXL-97兼容。
PDB2INS-用SHELXL 制备用于大分子精制的.ins和任选的 .hkl文件。对于已经存放在PDB中的结构,只需要四个字符的PDB代码。在PDB存款包含反射数据的大约95%的情况下,可以在不需要更改这些文件的情况下启动SHELXL细化。
CIFTAB和ShredCIF - 编辑和处理SHELXL的SM CIF文件。
SHELXC,SHELXD和SHELXE - MM定相。SHELXD对SM直接方法也很有用。
AnoDe - MM异常密度图的制备与分析。
使用说明
常用命令
一、选择原子或(Q)峰的方法
1.光标指在欲选择位置,点S键,可依次选择所有的要选择的原子或Q峰
2.光标指在欲选择位置,右手键点“Select”,可依次选择原子
3.用Select菜单(或背景右手键点“Select”)可选择部分和所有的原子或Q峰,顺序与原顺序相同
4.按住“Shift”键,左手键拖出一个选择框并同时轻开,可选择一个或多个原子
5.点refine菜单,系统会提示有Q峰没命名,并选择所有Q峰,顺序与原来顺序相同
二、删除原子或(Q)峰的方法
1.光标指在欲删除位置(原子或键),点K键
2.光标指在欲删除位置,右手键点Bonds and Angles,再在对话框中点“Delete”
3.光标指在欲删除位置,右手键点“Delete”
4.先选择,再点Select菜单中的Kill selected e在ins文件删除
三、恢复删错的原子或(Q)峰的方法
点“Edit>Restore Killed Atom”,先选择要恢复的原子(或Q峰),再点“Restore”
四、命名(标记)原子或(Q)峰的方法
1.先选择,再用Lablels菜单中的Group label
2.光标指在要命名的位置,用右手键的Edit
3.光标指在欲命名位置,右手键点Bonds and Angles,再在对话框中点“Rename”
常见问题
1、我收到错误消息**不允许ATOM NAMES **?
答:其中一条说明书中可能存在意外字符。查看.lst文件以查看导致问题的指令。角色可能是看不见的!
2、我收到消息** UNSET FREE VARIABLE FOR ATOM ... **但我没有使用任何'自由变量'?
答: 原子坐标中有一个拼写错误,例如缺少小数点或用逗号替换。或者你可能真的引用了一个未由a定义的 自由变量FVAR 指令!
3、我应该怎么做'可能被拆分'的警告?
答:可能没什么。只要有可能根据两个不连续的位置解释原子的各向异性位移,程序就会打印出这个警告,并估算这些位置的坐标。应该检查这些原子(例如借助于ORTEP图),但在许多情况下,单点各向异性描述仍然非常合适。
4、NPD是什么意思?
答:原子的U或Uij值已变为非正定。这可以通过以下方式预防XNPD 0.02在这种情况下,ADP-张量的特征值将被限制在0.02Å或更大,但找到原因可能更好。常见的原因是散射因子数被错误地指定,例如氧原子被氢原子散射因子精炼。另一个常见原因是两个原子(例如,无序成分)在(几乎)相同的位置上,但两者都没有EADP 约束或 SIMU 已采用约束来使其ADP值相等。 DELU 要么 日谷 当原子有不同时,不要这样做 部分 数字。
5、如何改进中子衍射数据?
答:插入一个NEUT 之前的指示 SFAC只使用元素名称的指令。这将使用中子散射长度,除了D之外,所有原子的自然丰度的所有同位素都是加权和,假设它是纯同位素。在解释说明时,它也会关闭H和D的特殊处理(例如ANIS 将使所有原子各向异性,包括H和D),但是 AFIX 说明书仍可用于放置和精炼氢。 FMAP -2可用于产生正和负'Q峰'。也可以看看HKLF 2 和 劳厄。
6、为什么来自SHELXL的CIF文件变得如此之大?
答:因为它们现在包含嵌入的.hkl,.res 和可能的.fab文件。这也是存档结构的好方法。如果您只打算使用.cif文件输入到另一个程序,则可以使用ShredCIF将其分解为其组件。这也可用于验证校验和以检查CIF文件是否已正确传输。
7、我是否应该使用TWIN和BASF进行最终细化以获得Flack x参数的确定值?
答:不!帕森斯的商法几乎总是给出更好的估计,甚至对于孪生结构也是有效的,但确实需要 (a)数据中存在弗里德尔对,以及(b) 结构已经完全改进。双胞胎(没有矩阵或具有负决定因素的矩阵)禁用帕森斯方法。有充分的理由相信基于Friedel对立面的商或差异的方法比在全矩阵细化中经典包含Flack参数给出更好的估计[Parsons,Flack和Wagner,Acta Cryst。 B69(2013)249-259]。
8、我的非中心对称结构没有比氧更重的原子,裁判坚持认为我使用的是MERG 4而不是MERG 2,但现在这似乎与ACTA不相容。我该怎么办?
答:阅读Thompson和Watkin,Tetrahedron Asymmetry 20(2009)712-717和Parsons,Flack和Wagner,Acta Cryst。 B69(2013)249-259,然后将你的论文发送给Acta Cryst。 代替!到目前为止,硬件和软件已经取得了进展,当最重的原子是氧时,即使用MoKα辐射,也很有可能确定绝对结构(至少有一种情况是纯碳氢化合物的CuKα数据)!因此,即使使用MoKα数据,最新的IUCr建议也永远不会平均弗里德尔对立面。这样做的另一个优点是可以获得更全面的数据统计(并写入.cif)如果使用了未合并的数据。通过不同地计算它们(Ton Spek建议)已经优雅地消除了参数esds中可能的人为减少的问题。MERG 2 对于大分子来说不是必需的 MERG 4 仍然可以用于他们,因为 ACTA 不需要指令,因为节省计算机时间更为重要。
9、我用小esd得到Flack参数0.5。我的结构是否与racemicaly结合?
答:可能不是!在许多这样的情况下,真正的空间群是中心对称的。外消旋孪生是可能的,但相对罕见。
10、我的结构只能在P1而不是P-1中解决,但在细化时,一些键长和U值对于两个分子是完全不同的。如果我使用相同的两个分子的几何形状变得相似,但我应该怎么做Uij?
A:你可以试试日谷,但寻找倒置中心可能会更好,否则你可能会被 摧毁!
11、如何从最终的.cif文件中生成漂亮的表 来填写我的论文?
答:运行CIFTAB并指定格式'rta'(Å)或'rtm'(SI单位)。这将生成一个Rich文本格式(rtf)文件,可以直接读入Microsoft Word或Open Office。然后将格式化表格,并且可以轻松地使用文字处理器添加个人触摸。然而,漂亮的图片甚至更好地填补了论文!
12、我怎样才能挤压我的结构?
答: “挤压”应理解为估算严重无序溶剂位点的复杂散射因子,并将其加入结构因子计算中。这种设施很容易被滥用,因此只有在有令人信服的理由时才应小心使用,例如因为不可能为沿结晶轴的无限溶剂密度的无限河流定义原子模型。程序PLATON能够计算用于输入SHELXL的适当的部分结构因子ABIN 指令。 ABIN还规定了可以针对部分结构因子进行细化的比例因子和总体B值,并且还可以用于孪晶结构,在这种情况下,部分结构因子应该对应于未淬火结构。有关详细信息,请参阅 PLATON文档。
13、我已经通过MR解决了我的结构,但我无法取得进一步的进展。我该怎么办?
答: 如果您足够幸运,具有良好的分辨率(2.5A或更高)和高溶剂含量,检查解决方案是否正确的简单(但缓慢)方法是将PDB文件从MR重命名为name.pda 和把它喂给SHELXE:
shelxe name.pda -s0.55 -a20 -q -t4
哪里 -s设定溶剂含量。如果针对本机数据的CC上升到25%以上,结构几乎肯定会被解决,name.pdb中的poly-Ala跟踪可以帮助您解释它。在边缘情况下的新选项-o 和 -O在SHELXE中可以通过优化起始模型来提供帮助。如果异常数据也可用,MRSAD可以改善阶段,例如
shelxe name.pda name_fa -h -z -q -a5 -t4
在这种情况下, 应首先运行SHELXC(或hkl2map)以生成文件name_fa.hkl。
14、使用SAD或MAD方法输入强度数据以解决结构的最佳方法是什么?
答:在hkl2map中 输入XDS_ASCII.HKL,name.sca或name.hkl类型文件。如果你处理与XDS数据,这是 很多更好的输入XDS_ASCII.HKL文件,而不是直接打伤数据与第一其它程序!以这种方式使用未合并数据也可以提供更好的统计数据,例如CC1 / 2。此文件可以重命名,例如重命名为XDS_PEAK.HKL,XDS_INFL.HKL和 XDS_HREM.HKL,用于典型的MAD体验,因为无论调用什么,SHELXC都会识别它。如果您使用的是布鲁克系统,最好使用未合并的 .hkl文件。Bruker XPREP可以读取,写入和转换各种格式,将数据集缩放在一起,确定空间组并将单元和数据转换为适当的常规设置,并添加或传输free-R标志。它还提供了SHELXC的替代方法,用于制作 name_fa.hkl和.ins文件。
15、为什么硫磺-SAD不能成为分阶段新蛋白质结构的标准方法?
答: Sulfur-SAD需要非常准确的数据。许多光束线不够稳定,不足以产生这样的数据,辐射损伤很容易淹没1-2%的异常信号。使用kappa或三圆测角仪收集的高度冗余的内部数据通常可以提供更好的相位信息,但将其与更高分辨率的原生同步加速器数据(不需要如此高的精度或冗余度)相结合仍然可以对地图产生重大影响质量和自动跟踪的便利性。
16、如何微调子结构解决方案?
答:改变分辨率截止值,进行更多试验(一些子结构在50000次试验中仅解决一次),使用二硫化物选项(DSUL)在SHELXD中尝试不同的空间组。如果您收集了MAD数据,请尝试使用SMADSHELXC中的选项使用SAD的组合数据。新的-z 强烈建议在SHELXE中选择优化和扩展子结构。
17、如何使用SHELXE微调相位?
答:改变溶剂含量(-s),密度修改周期数(-m),包括特定的α-螺旋搜索(-q并给它更多的时间进行螺旋和肽搜索(例如 -T4)。然而,就CPU时间而言,最后两个选项是昂贵的。如果您有可能的搜索片段和弱异常数据,MRSAD(参见Q20)可能会有用。
18、如何更好地了解SHELXE跟踪和地图?
答:用添Grüne的shelx2map的转换.phs成CCP4格式的地图文件,然后输入该文件和.PDB从SHELXE到PYMOL。您可能还希望添加例如-e1.0到SHELXE命令行,使用免费午餐算法改进最终地图。
19、为SHELXL 创建.hkl输入文件的最佳方法是什么?
答:许多商业系统(例如Bruker APEX2 / PROTEUM2)可以直接创建此文件。如果您使用SHELXC / D / E或hkl2map来解决结构,则会将此文件作为副产品获取。如果你只有.mtz文件,请务必保持强度并在CCP4中设置free-R标志。然后你可以使用TimGrüne的mtz2hkl制作.hkl文件。如果.mtz文件中没有强度 ,mtz2hkl可以生成包含F值而不是强度的.hkl文件,但是在读取文件时必须小心指定。另见Q21。
20、如何为SHELXL 设置第一个.ins文件?
答:您必须使用SHELXPRO(SHELX-97的一部分)中的“I”选项,直到更好的程序准备就绪。然后,您需要自己添加除20种标准氨基酸以外的残留物的限制。
21、我在哪里可以找到合适的几何约束?
答:一个好的开始是CCP4单体库。更好的是,Dale Tronrud设置了一个 cdl_shelxl Web服务器,可以读取PDB文件并输出改进的构造依赖DFIX, 党 和 CHIV限制SHELXL蛋白质的改良。有关详细信息,请参阅Tronrud和Karplus, Acta Cryst。 D67(2011)699-706。对于小分子, PRODRG Web服务器 可以生成SHELXL格式的几何约束。Global Phasing GRADE Web服务器可能提供最准确的约束,也可以直接以SHELXL格式创建它们(DFIX, 党 和 平面)。这些约束可以插入 .ins文件或写入单独的文件,并使用SHELXL .ins文件中的'+ filename'或'++ filename'选项读入。
22、如何在SHELXL细化后观察电子密度?
答:运行SHELXL细化列表6,然后较新版本的Coot将能够读取生成的.res和.fcf文件并显示地图。不幸的是,如果您使用Coot编辑模型并尝试将其写入.ins文件以进行下一个SHELXL细化作业,则几乎肯定需要进行一些手动编辑。TimGrüne的shelx2map可用于将.fcf文件转换为PYMOL可以读取的CCP4格式映射(以及来自SHELXL 的.pdb文件)。这是获得包含电子密度的出版物质量图的推荐方法。
23、我如何计算标准偏差?
答: 这需要全矩阵细化。通常只需要坐标中的esds和派生的几何参数。精炼后与之融合CGLS,单周期的全矩阵细化(LS 1)仅针对坐标执行(BLOC 1),删除所有几何约束和零移位乘数(DAMP 0 0)。可能需要微调内存分配-一个 和 -b运行时标志。对于非常大的结构,可能需要使用由定义的重叠块进行几个细化周期集团说明。为了获得各种几何参数的esds,考虑完整的相关矩阵, RTAB, MPLA, 键 和 HTAB可能用过了。.lst文件中原子名称下面出现的数字是Ångstroms中的径向原子位置esd。
24、如何在对称元素上生成约束?
答:您需要指定一个EQIV 指令,然后参考对称生成的原子使用 _ $ 1 例如,如果Cys29的SG与其对称等效物的二硫键被(0.5,y,0.5)的晶体双轴一分为二,则需要指定:
EQIV $ 1 1-x,y,1-z DFIX_29 2.031 SG SG_ $ 1 DANG_29 3.035 CB SG_ $ 1 SG CB_ $ 1
25、我什么时候应该加氢?
答: 使用添加氢HFIX 要么 AFIX改进了模型,不添加任何额外的参数。最好在添加氢之前对模型进行模拟,因为程序将正确添加无序氢(!)。然而,不建议在-OH基团(Tyr,Ser和Thr)中加入氢, 因为(a)它们对R值几乎没有影响,(b)程序难以准确预测其位置, (c)如果程序意外地将两个氢分配到相同的H键,然后使用骑行模型进行精炼 (AFIX 83)打开防撞击限制(BUMP),氢之间的排斥可能会在结构中引入机械变形。应将氢单独添加到组氨酸的环N原子中,考虑可能的H-键和金属配位(或省略)。向蛋白质中的C和N原子添加氢通常会使R1和R1(游离)减少相同的量,通常为0.5至1%。使用精确的小分子数据,下降可以大得多。
26、当我添加氢时,程序说:
** BAD AFIX CONNECTIVITY:CB_21 BONDS TO CA_21 **。我该怎么办?
答:该计划无法将氢添加到CB_21,因为侧链的其余部分缺失。当侧链的其余部分没有密度时,这是推荐的过程,但是有必要添加一条指令:HFIX 0 CB_21 在冒犯之前 HFIX指示关闭仅为此特定残留物添加CB氢。类似的错误消息也可能由不良几何导致连接数组中的错误导致,这些可以通过更正自由 和/或 BIND 说明。
27、加入氢后,SHELXL抱怨:
** BAD AFIX连接:N_1 BONDS到CA_1 **。怎么了 ?
答:该计划正试图将终端N变成酰胺而不是-NH3+,它不起作用,因为它只与一个原子键合。包括HFIX 33 N_1 在另一个之前 HFIX说明。该程序始终适用于第一个 HFIX 这适合于给定的原子。
28、C末端是否需要特殊限制?
答:是的。通常C-末端是羧酸盐(-CO2-)具有相等的CO键长度,如Glu或Asp侧链的末端。然后最好将这些原子命名为OT1和OT2(以便不应用通常的限制)并添加适当的额外限制,例如
DFIX_129 1.249 C OT1 C OT2 DANG_129 2.194 OT1 OT2 DANG_129 2.379 CA OT1 CA OT2
29、模拟无序侧链的最佳方法是什么?
答:用Coot看地图,看看哪个是第一个无序原子(比如CG)。然后返回一个原子(到CB)并放一个 第1部分10.66 在它和之前 第0部分在侧链的最后一个原子之后。运行另一个改进工作并再次查看密度,第二个构造现在应该更加清晰,并且应该可以将它与Coot拟合。您可能必须移除一个或两个试图模仿第二个构造的水。添加 0.66 到最后 FVAR 为第一个组件的占用引入一个新的自由变量的指令(比如fv(5)),改变 部分 第一个组件之前的指令 第1部51 并插入一条指令 第2 -51部分在第二个组件之前(也应该从CB开始)。然后,第二组分中的原子都具有1-fv(5)的占有率。fv(5)将从0.66的起始值进行细化,这可能是一个很好的第一近似值。在以正常方式添加氢之前,最好这样做HFIX这样就会自动考虑到这种疾病。通常,原子在不同部分 应该具有相同的名称,并且约束将适用于两种构造 部分考虑到了。如果两种(或更多种)组分在化学上不同,则应给它们不同的名称。然后可以使用这些名称为仅一个组件添加适当的约束。
30、我有一种环状蛋白,其中N_1与C_99形成正常的肽键。我如何说服该程序将肽限制应用于这些残留物?
答:只需改变 RESI 残留物1的说明(比如说 RESI遇到1) 至 RESI遇到1 100。第二个残留数是别名。该_ + 和 _-连接相邻残基中原子的束缚使用的算子适用于残基数及其别名(如果有的话)。相同的技术可用于桥接序列中的缺失; 插入是可能的,但更棘手(不仅仅是SHELXL!)。
31、Rfree在各向异性上仅下降0.2%,但地图要好得多。当然我应该继续各向异性细化?
答:不!回到各向同性。Rfree下降0.2%通常不显着,因为Rfree使用相对较少的反射。保持R和Rfree之间的差距尽可能小也很重要。你的地图看起来“更好”,因为它们看起来更像你的模型 - 这就是过度拟合(或模型偏见)的全部意义!
32、是否有可能读取部分结构因素,例如,对于比SHELXL提供的原始Babinet模型更复杂的溶剂模型?
答:是的。使用ABIN读取包含h,k,l 的.fab文件以及计算出的结构因子的实部和虚部,以获得更好的溶剂模型(您可能需要编写一个小程序来生成此文件)。ABIN允许为这些贡献细化单独的比例和B因子。这甚至适用于孪生结构(.fab文件应仅针对一个组件计算)。
33、如何生成欧米茄角的直方图?
答:一个实用的解决方案是在最后一个.lst文件中取出“二面角OMEG”后面的表格,将其 粘贴到另一个文件中,然后使用gnuplot或某些Office软件制作直方图。不是很优雅,但它的工作原理。
34、Molprobity和PDB不喜欢SHELXL的氢原子名称,并抱怨-CH的“手性”2- 团体!
答:没有氢的 沉积结构 (WPDB 2)!如果您需要将氢用于其他目的,可以使用MolProbity以常规名称再生它们。SHELXL不喜欢以数字开头的原子名称,因此不能使用这些原子名称。
35、Coot可以正常使用SHELXL-97 的.fcf文件,但无法读取新SHELXL中的文件!?
答: IUCr CIF委员会弃用了'_symmetry_equiv_pos_as_xyz'并将其替换为'_space_group_symop_operation_xyz',因此必须在.cif和.fcf文件中使用新名称。许多读取这些文件的程序(包括Coot,CIFTAB / XCIF,PLATON,CheckCIF等)都被作者迅速修改,以了解这两个名称。当然,较旧版本的Coot等不了解新名称。请下载最新版本的Coot,这个问题就会消失。这只是CIF定义不必要的变化导致的巨大混乱和浪费时间的一个例子!