COSMOthermx18破解版是一款用于计算液体的热物理数据的软件程序。安装完成后还包括COSMObaseEditor18和COSMOview18两个可独立运行的程序,COSMOtherm计算的一般过程包括化合物选择和属性输入两个步骤,并具有图形化的界面,COSMOtherm计算需要筛选涉及系统的分子的电荷分布。筛选电荷分布可从分子的筛选电荷表面获取,如量子化学COSMO计算中所计算。 COSMO计算的结果存储在扩展名为.cosmo或.ccf的COSMO文件中。选择化合物后即可转到属性选项卡选择要计算的属性,属性面板包含丰富的可调整设置内容,非常的灵活。软件支持同时运行多个作业,作业完成后结果可在窗口中单独显示。COSMOtherm具有批处理功能,以及数据库处理和搜索功能,适用于许多应用的可压缩液体和天然气,可从根本上修订和改进的许多属性算法以及高质量预测级别BP-TZVPD-FINE的一些改进等,使用软件,您将能够轻松的进行流体数据的计算和预测,从而快速的了解和验证各种材料的属性,软件界面简单直观,具有强大的性能和高效的流程,让您几秒钟就能够看到详细的结果,本次带来破解版下载,含破解工具,有需要的朋友不要错过了!
安装破解教程
1、在本站下载并解压,如图所示,得到COSMOthermX_windows-x64_18_0_1_Java8.exe和crack破解文件夹

2、双击COSMOthermX_windows-x64_18_0_1_Java8.exe,勾选我接受许可证协议,点击next
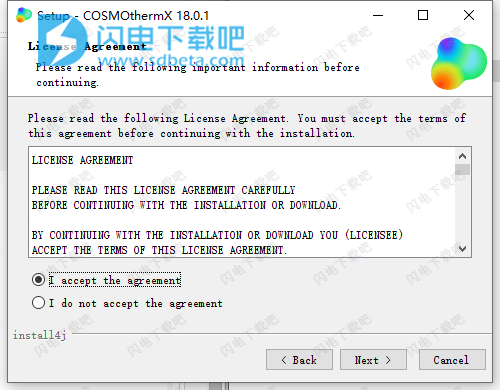
3、选择软件安装路径,点击next
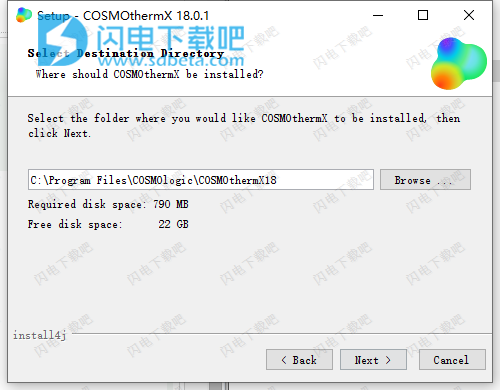
4、安装需要稍等一会儿,如图所示,安装完成,点击finish
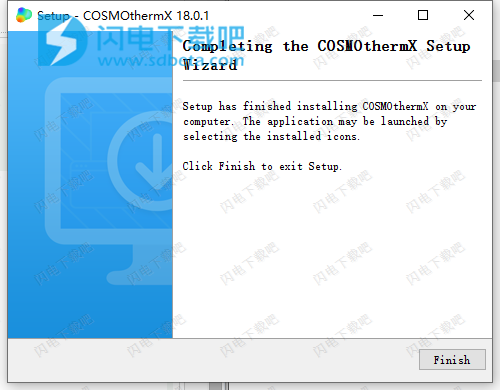
5、安装完成,管理员身份运行COSMOlogic_Patch.exe,如图所示,选择安装目录,点击start
功能特色
1、集成在该软件的设计和设计中
2、一个非常用户友好的GUI,使用户可以轻松完成任务
3、可以执行高精度预测
4、特别是减少了计算和预测中的消耗时间
5、高处理速度意味着几秒钟内的答案和结论
6、预测液体,固体和气体溶剂状态的物质的行为
7、活动活动预测,两相分散(例如LogP)
新功能介绍
1.新的,扩展的和修订的图形用户界面CoSMOthermX功能批处理:新的图形用户界面支持分子批次的提交,从而可以计算一系列分子的特性。
远程提交作业:现在可以在remotelinux服务器上提交COSMOtherm计算。这使用户可以轻松地处理许多或非常耗时的工作。
项目树。现在可以在项目中处理作业,因此可以更轻松地进行管理。
新的数据库结构:现在可以通过SQL数据库交付和处理预安装的数据库和COSMObase。访问速度更快,并且可以在数据库搜索中实现其他功能。较早的COSMOtherm版本格式的数据库可以转换为新格式。
优化的数据库界面:搜索新分子变得更加容易,快捷和强大。现在可以将分子名称,CAS号甚至SMILES粘贴到窗口中,并在几秒钟内检索所有分子。
其他更改:FlatSurf和“界面张力”面板已收缩到“界面属性”面板中。其他更改包括“相似性”,“多种溶剂”和“ COSMOmic”面板。
2.新的,扩展的和修订的coSMOtherm功能改进的化合物装载:由于COSMOtherm代码的更改,分子的装载速度更快,从而加快了包含许多分子或一系列小任务的工作。
新功能:COSMOtherm中引入了所谓的“混合化合物”(混合文件)的概念。 MIX文件概念允许复合材料的简单应用
(例如离子液体,盐或与结晶水形成的盐)或具有固定混合比的混合物(例如盐水,这是具有固定组成的水中的岩盐溶液)。这样定义的伪化合物可以在COSMOtherm的工作流程中像单个常规化合物一样对待。
这样可以大大简化具有固定混合比的复合材料和混合物的处理。而且,现在在所有COSMOtherm功能和特性预测面板中通常都可以使用这种特殊化合物的处理方法。混合化合物或复合材料的源文件(即混合化合物成分的COSMO,ENERGY和VAP文件)内部存储在MlX文件中,因此避免了任何兼容性和歧义性问题。请注意,此功能在图形用户界面CoSMOthermX中尚不可用。
新功能:将COSMOtherm气相相关性质的预测(当前的蒸气压,沸点,汽化焓,亨利定律常数,溶剂化的自由能,密度)扩展到临界条件。这是通过将常规COSMO-RS理论(基于不可压缩的液相和理想气相的假设)与状态方程(EoS)方法及其相应的混合规则(MRs)相结合而实现的。注意,此功能在图形用户界面COSMOthermX中尚不可用。
新功能:扩展了COSMOtherm VLE和LLE相图预测。现在可以计算出等压或等温相图的温度或压力函数。滑移函数假定要固定的气相y中的摩尔分数浓度和液相y中的浓度是y的因果函数。因此,与常规默认VLE和LLE计算相比,glide选项基于不同的支点来计算VLE和LLE相位图。注意,此功能在图形用户界面COSMOthermX中尚不可用。
增强功能:扩展了天然COSMOtherm密度和摩尔体积预测,并具有温度和混合物依赖性。在COSMOtherm的早期版本中,此功能仅适用于室温下的纯化合物。与旧模型相比,现在已经扩展了密度模型,以预测在任何温度p(T,)下混合物的密度,而不会损失预测质量。注意,此功能在图形用户界面COSMOthermX中尚不可用。
增强功能:扩展了具有温度依赖性的本地CoSMOtherm粘度预测模型。与旧的仅室温模型相比,现在已经扩展了粘度模型,以预测纯化合物在任何温度n(T)下的粘度,而不会损失预测质量。注意,此功能在图形用户界面COSMOthermX中尚不可用。
新功能:已开发并实现了扩散系数(DC)预测模型。 D(T,x)模型适用于纯化合物的自扩散,在溶剂中无限稀释的化合物以及在任何温度下的混合物。注意,此功能在图形用户界面COSMOthermX中尚不可用。
新功能:开发并实施了一种预测液体导热系数(LTC)的模型。 k(T,x)模型适用于纯化合物,并且适用于任何温度下的混合物。注意,此功能在图形用户界面COSMOthermX中尚不可用。
新的和扩展的功能:一般已实现了温度序列(用于等温固定压力特性)或压力序列(用于等压固定温度特性)的计算,现在可用于COSMOtherm中可用的所有特性计算。现在,除了输入一系列温度或压力步数外,还可以给出温度或压力增量。请注意,此功能在图形用户界面COSMOthermX的所有面板中尚不可用。
新功能:输入实验温度相关的纯化合物密度,以用于聚合物预测。可以通过DIPPR105方程,DIPPR116方程或多项式展开的参数输入rexp(T)。另外,可以输入密度和温度对,这将由COSMOtherm拟合到多项式。实验密度值可用于聚合物预测所需的自由体积计算中。
注意,此功能在图形用户界面COSMOthermX中尚不可用。
修订和改进的功能:界面张力(IFT)预测和用于计算表面自由能属性的Flatsurf功能已组合在一起,以更易于使用,并重新实现为COSMOtherm的命令行版本。因此,现在也可以在COSMOtherm的命令行版本中计算IFT。
修订和改进的功能:溶解度多重溶质和多种溶剂的筛选已统一在一起,易于使用,并重新实现到了COSMOtherm的命令行版本中。
因此,现在也可以在命令行版本的COSMOtherm中计算多种溶剂溶解度筛选。此外,参考溶解度功能已进行了根本性的修订,现在也可用于多种溶质和多种溶剂的筛选。
修订和改进的功能:离子液体容量和选择性筛选(IL筛选)选项已统一,便于使用,并重新实现到COSMOtherm的命令行版本中。因此,现在也可以在COSMOtherm的命令行版本中计算IL筛查
修订和改进的功能:COSMOmeso功能(即将COSMOtherm计算的活动系数与Flory-Huggins和DPD模拟参数的拟合)已统一以便于使用,并重新实现为COSMOtherm的命令行版本。 因此,现在也可以在COSMOtherm的命令行版本中计算COSMOmeso参数。
3.提高了coSMOtherm预测的准确性和适用性我们的“最佳质量”计算级别BP-TZVPD-FINE理论得到了进一步增强。
当前的BP-TZVPD-FINE级别包括以下功能:
·改进的氢键结合可降低噪声至极端o值(2018年3月)
强大的网络溶剂的新的焓贡献,即水(2017年12月)
氢键校正错配(HBCMF)(2016年12月)
残留介电校正(RDC)(2014年12月)
基于Grimme D3方法的色散能(2013年12月)
HB相互作用能的色散校正(2013年12月)
HB受体位点的熵贡献(2012年12月)
新型氢键物理(2011年12月)
氢键的位阻(2011年12月)
包含一些氢键键合效应(2011年12月)
·新的量子化学能级(BP-TZVPD)(2011年12月)
·改进型腔结构(细腔)(2011年12月)
参数集改进:增强了COSMOtherm参数集,可实现高水平(BP-
TZVPD-FINE),生产水平(BP-TZVP-COSMO和DMOL3-PBE)和筛选水平(BP-SVP-
AM1)的sigma-profiles是通过仔细考虑拟合数据和对涉及拟合和验证过程的所有化合物的构象异构体选择而获得的。
由COSMOtherm计算的化学势的参考状态已被修改。
现在,将液态和气相的化学势与物理上有意义的数字进行比较。请注意,这种变化不会影响化学势的差异。 COSMOtherm计算出的所有可观察特性,以及所有不能直接测量但基于化学势差的特性
(例如活度系数或溶剂化自由能)将不会因参考状态的变化而受到影响。
使用帮助
一、缩略语
2D:二维
3D:三维
AM1:半经验量子化学方法
BP-SVP / BP-TZVP:Becke-Perdew1、2、3(BP)用于密度泛函理论计算,具有分裂价加极化函数(SVP)或三价加极化函数(TZVP)基础集。必要的参数化文件始终始终与一个功能和基础集相对应。因此,有时会使用术语“ BP-TZVP参数化”,它是指COSMOtherm参数化而不是基础集规范。
CAS号:化学文摘社注册号是化合物的唯一标识符。
COSMO:类似于屏蔽模型的导体
COSMO-RS:像真正的溶剂筛选模型一样的导体
DB:数据库,通常用于COSMOtherm复合数据库。
DFT:密度泛函理论:一种在多个软件包中用于分子或晶格计算的量子化学理论。
HB:氢键
IL:离子液体
LFER:线性自由能关系
LLE:液体-液体平衡
MW:摩尔重量
QC / QM:量子化学/量子机械
QSPR:定量结构性质关系,也称为QSAR(A =活性)
(sigma):COSMO筛选费用
SLE:固液平衡
微笑:简化的分子输入线输入规范,用于描述分子的符号,例如甲烷是“ C”,乙烷是“ CC”,乙醇是“ CCO”
SMS:西格玛匹配相似度
VLE:蒸气液体平衡
二、化合物
COSMOtherm计算需要筛选涉及系统的分子的电荷分布。筛选电荷分布可从分子的筛选电荷表面获取,如量子化学COSMO计算中所计算。 COSMO计算的结果存储在扩展名为.cosmo或.ccf的COSMO文件中。为了在COSMOtherm中使用,必须在可使用COSMOtherm参数化的量子化学级之一上生成COSMO文件。对于标准化学和工程热力学中的应用,我们建议使用TZVP或TZVPD-FINE水平。这两个级别都基于从DFT计算中检索到的分子结构,但是TZVPD-FINE级别涉及带有TZVPD基础集的其他QM能量计算。通常,在TZVP级别上的COSMOtherm计算速度更快,而TZVPD-FINE级别给出的结果略好。根据所涉及化合物的数量,可用的计算机功能以及所需的精度,其他级别可以更好地适合于其他目的。 COSMOtherm从化合物的COSMO文件中读取化合物信息,并将筛选电荷表面转换为筛选电荷分布,称为分布。通常不需要有关分子结构的信息并将其丢弃。由于一种化合物的构象异构体的-分布可能会因分子结构而异,因此必须分别计算构象异构体。如果某个化合物存在多个构象异构体的COSMO文件,
它们将合并为一个化合物。有关选择用于COSMO计算的构象异构体以及COSMOtherm中的构象异构体处理的详细信息,请参阅第5.3节。在启动时,COSMOthermX工作场所窗口中有两个选项卡:COMPOUNDS和PROPERTIES。可以从化合物选项卡中选择化合物。有两种方法可以执行此操作:从数据库之一中,选择所需级别,然后单击FROM DATABASE按钮。 在计算机系统上的目录中,单击“从文件”按钮。要从数据库之一中选择化合物,请选择一个级别,例如TZVP,然后打开列出所有可用化合物的数据库。数据库对话框具有多种功能:默认情况下,将列出所选级别的所有嵌入式数据库中的化合物。可以使用相应的复选框来选择或取消选择数据库。鼠标悬停将显示数据库目录的位置。 可以通过选中第一列中的框来标记化合物以供选择。当按下添加到化合物列表的添加按钮或添加选择和关闭按钮时,所有标记的化合物将被转移到主窗口中的化合物部分。 双击化合物也将化合物转移到主窗口中的“化合物”部分。 单击列标题将根据该列对数据库进行排序。 默认情况下,将选择化合物的所有构象体。如果取消选中“使用所有符合要求者”复选框,则仅最低能耗的符合者将被转移到复合部分。 如果需要除最低能量协调器之外的特定协调器,请首先选择完整的协调器集。关闭数据库对话框,并突出显示“化合物”部分中设置的构象体。使用鼠标右键打开复合上下文菜单,然后选择DECOMPOSE CONFORMER SET。然后通过突出显示它们并按“ Del”键,从复合部分中删除不需要的构象异构体。 数据库表是可搜索的。输入搜索字符串(名称,公式,CAS号,微笑代码),然后按SEARCH或SEARCH SMILES处理搜索。
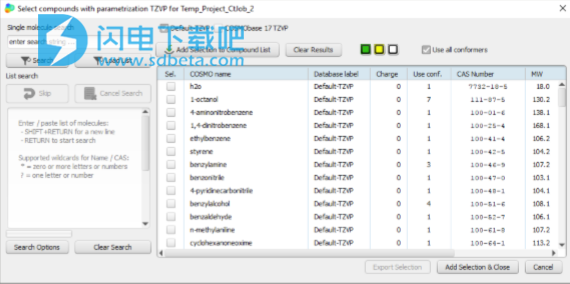
要从目录树中的某个位置选择化合物,请单击“文件管理器”按钮。 转到所需化合物的COSMO文件所在的目录,并突出显示这些文件。 单击文件名时,按住“ Ctrl”或“ Shift”键可以突出显示几个文件。 要将突出显示的COSMO文件传输到复合区,请按FILE MANAGER窗口左侧的SELECT按钮。 如果无法从COSMO文件中识别出参数化,则将要求您选择参数化以适合所选文件。 确保使用适当的参数化,即与量子化学COSMO计算中使用的DFT功能和基础集相对应的参数化。 所选化合物列在“化合物”部分中。
三、属性输入
选择化合物后,转到“属性”选项卡,然后选择要计算的属性。每个属性面板均包含可调整设置的字段。所有属性计算所需的设置是温度和系统组成。可以通过三种方式设置系统的组成:如果它仅由一种化合物组成,只需在PURE列中的方框中打勾即可。如果系统由几种化合物组成,则摩尔数或质量分数数可以在相应的字段中进行复合设置。或者,可以使用复合线中的滑块设置摩尔分数或质量分数。根据属性,其他输入,例如可能需要第二阶段的组成。设置完所有参数后,按输入准备面板底部的ADD按钮以完成属性输入。可以为单个作业中的多个COSMOtherm计算准备输入。要运行作业,请按“运行”按钮。可以保存作业并使用作业名称运行。如果未使用作业名,则作业将作为临时作业运行,并且在关闭COSMOthermX时会删除输入和输出文件。在使用“另存为”按钮运行之前,作业输入文件也可以永久保存。
四、工作场所
可以在主机内部打开多个工作场所窗口。 可以同时设置和运行多个作业。 作业完成后,计算结果将显示在工作场所窗口的单独选项卡中。 要打开一个新的工作场所,请使用“文件”菜单中的“新建作业”,或直接单击工具栏中的快捷方式图标。 如果将视图更改为CLASSIC STYLE,则作业列表将最小化到工具栏中。 工作区窗口被放大以覆盖主窗口,并且COMPOUNDS和PROPERTIES选项卡并排布置。
捐助666获取,送本站会员