
安装破解教程
1、在本站下载并解压,在本站下载并解压,得到以下文件
2、双击TmoleX42_Win64_DEMO.exe运行安装,如图所示,稍等一会儿
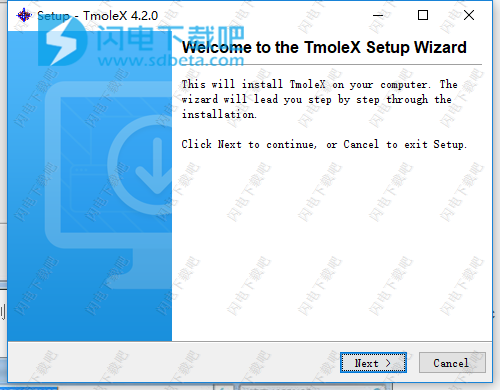
3、点击next
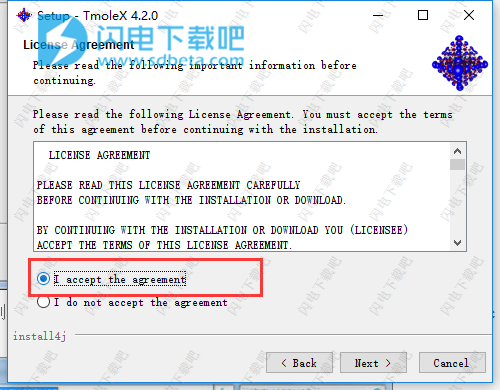
4、勾选我接受协议选项,点击next
5、点击浏览选择软件安装路径,点击next
6、安装中,稍等一会儿
7、选择检查更新,点击next
8、点击finish退出向导
9、不要运行软件,将安装包中的turbo_license.ctd复制到COSMOlogic \ TmoleX16 \ TURBOMOLE \ LICENSE文件夹中
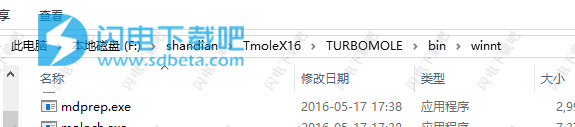
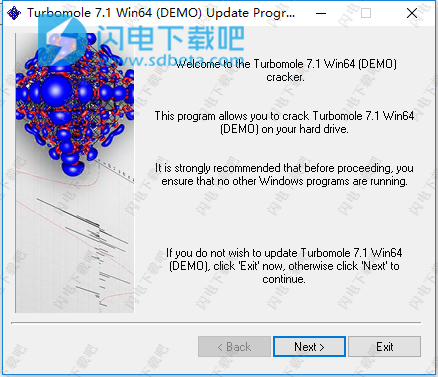
10、将TM7.1Win64-patch.exe复制到COSMOlogic \ TmoleX16 \ TURBOMOLE \ bin \ winnt中运行,如图所示,点击next
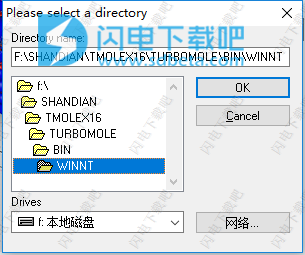
11、如图所示,点击...打开并直接双击最下面的winnt,然后点击ok
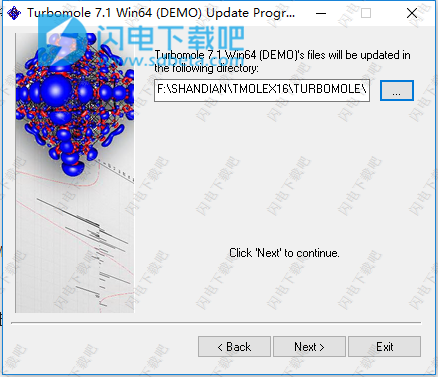
12、点击Start按钮并稍等一会儿
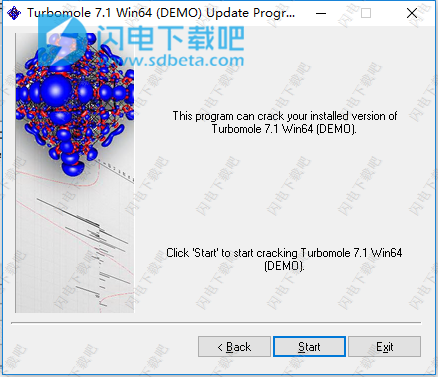
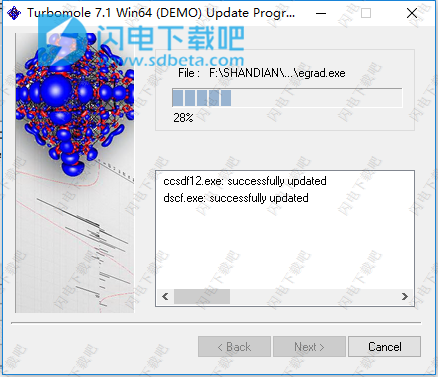
13、完成,退出向导
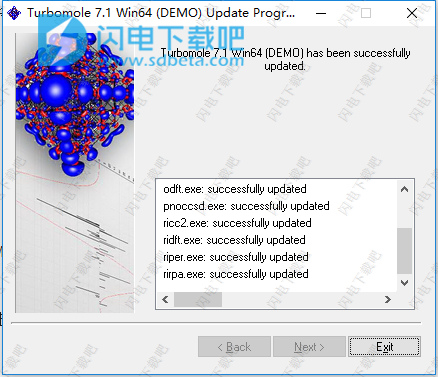
14、如果需要进行多核计算,请在COSMOlogic \ TmoleX16 \ TURBOMOLE \ bin \ winnt_smp中运行TM7.1Win64smp-patch.exe
使用帮助
如何创建输入如果TuRBoMoLE位于/ my disk / my name / TURBOMOLE,则可以通过在(类似bash)shell中执行以下三个命令来访问所有脚本和程序:export TURBODIR = / my disk / my name / TURBOMOLE export PATH = STURBODIR / scripts:SPATH export PATH = STURBODIR / bin /“sysname:SPATH
坐标
首先,您需要分子的3D结构:
1.构建TURBoMoLE模块,以交互方式生成输入(详细信息如下所述),定义,具有一些有限的功能,从头开始构建结构,但这既不方便也不直观,除了一些小的情况,特别是如果你应用对称性。图形用户界面TmoleX包含分子构建器,可从COSMOlogic网站免费获得。
1.使用TmoleX导入结构,修改它们或从头开始构建,为TURBOMOLE定义完整的输入,将作业提交到本地机器或远程系统,获取结果并分析数据。
2.使用外部构建器(Molden,ECCE,Hyperchem,Insight,Arguslab,MAPS,Accelrys DS Visualizer ....
有很多!)并将结构转换为xyz格式。
3.从数据库或Internet获取结构,并通过您选择的定义或构建器对其进行修改。
2.转换使用TURBoMoLE脚本x2t将xyz文件转换为TURBOMOLE坐标:x2t struct。 xyz> coord脚本计算可以读入xyz,sdf,ml2,car和cosmo文件,而TmoleX能够导入各种坐标类型。
从结构到计算
有几种方法可以获得完整的控制文件并进行计算。这些是最广泛使用的:
1.定义a)运行定义b)运行任何TuRBoMoLE模块(程序或脚本)。
您必须从基态能量计算开始,以获得带有dscf或ridft的收敛轨道
(例外:jobex默认执行初始能量计算)。
2.计算生成*。 COSMOtherm输入的cosmo文件。
a)制作分子列表b)通过使用脚本计算,在所有给定分子上批量运行COSMO计算
3. Tmolex TmoleX是COSMOlogic的TURsoMoLE的免费附件。用于Linux / PC,MacOSX和Windows的版本可从通常的TussoMoLE发行版所在的ftp服务器获得(请咨询您的TURBOMOLE管理员)。还有一个免费的客户端版本,可用于构建分子,生成输入文件并将TuRsoMoLE作业提交给远程计算机。有关客户端版本的详细信息,请查看COSMOlogic主页。
TmoleX可以导入坐标,更改结构,生成输入文件和运行TURBoMoLE作业。结果显示在TmoleX内的小组中。可以显示结构,几何优化和振动频率并设置动画。请访问网站:TmoleX,Turbomole GUI
快速浏览如何运行计算
创建输入:最短的定义方式正如我们在前面的章节中所看到的,有一个特殊的程序可以创建一个完整的TuRsoMoLE输入:define大多数定义的功能都不是在你的日常工作中需要。通常,按<ENTER>将打印出当前菜单,*将继续执行下一步。大多数情况下,您可能只接受所有默认值。在前三个菜单中,加载几何图形,分配基础集,并创建起始MO和占用。如果您在最后一个菜单中没有更改任何内容,您将获得一个很好的Hartree-Fock输入。在最后一个菜单中输入dft,ri,mp2,cc,...将打开或关闭其他方法。
因此,输入的最短路径是获取TURsoMOLE坐标(文件coora)或xyz文件中的结构
(struct.xyz)然后让Turbomole的x2t脚本将其转换为coord文件:x2t struct。 xyz> coord然后创建一个新的空目录并在那里复制coord文件。切换到该目录并调用define。首先,系统会要求您输入输入文件的名称,只需在此处按<Enter>键,然后输入标题即可。标题可以只是一个空行(在这里给出标题可能是一个好主意,有时候是查看输入内容的最快方法)。
这里解释了define中最重要的命令。 *表示四个主菜单中的每一个都结束而下一个菜单开始的位置,因为*或g是结束菜单并继续下一个菜单的命令(见下文)。
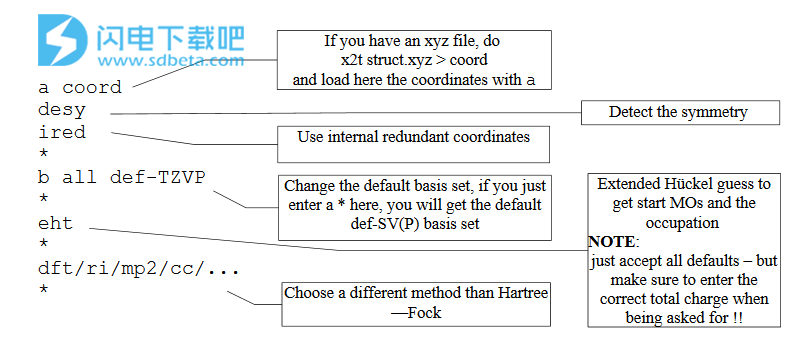
之后,您有一个很好的Turbomole输入,您可以运行任何Turbomole模块或脚本 - 具体取决于您要计算的属性。
创建输入并运行计算第一个练习!
我们只是从TURBoMoLE结构库中获取其中一个结构,而不是从xyz文件开始,这是每个TuRBoMoLE发行版的一部分。 我们将为几乎所有其他后续的例子做到这一点......
1.使用define创建输入:
·从新目录或空目录开始,调用define并输入以下内容:
<回车>
这是第一个练习,我们在苯上进行HF计算! 苯desyired eht
<回车> <回车><回车>
上面给出的所有命令将在本教程的后面解释。 (几乎)相同的练习在第6.1章中进行 - 详细描述了输入和输出。 如果您对此示例有困难,请尝试第6.1章!
一些注意事项:当然,您可以跳过冗长的标题,或者只需按<Enter>键即可完全不提供标题。
结构库中的分子像任何其他坐标文件一样被加载,唯一的区别是有一个感叹号'!'命令a和分子名称之间。
我们让定义用desy确定对称性,并定义找到的Doa点组。
使用ired自动确定内部冗余坐标 - 这里不需要这样做,但是如果未调用ired,则会在退出几何菜单(第一个*)时询问您是否确定不使用内部坐标。
我们接受了默认基组def-SV(P) - 这是TURsoMoLE默认(def-)基组之一,SV(P)
是一个双zeta价基组,在所有非氢原子上都具有极化。
eht运行扩展的Hickel计算以创建起始轨道并确定占用/多重性,其余的<Enter>键属于Hickel要求您的问题。请注意,其中一个问题是分子的总电荷(默认为0-中性),另一个问题是如果您接受找到的职业
(闭壳系统,在这种情况下是微不足道的)。
最后,最后的*退出定义,在最后一个菜单中没有任何改变 - 因此我们有一个标准的Hartree-Fock
2.运行Hartree-Fock能量计算:dscf> dscf。 计算完成后(大约需要3秒钟),请查看输出文件dscf。 out:你会看到9次迭代已经完成,总能量是-230.51807365370 Hartree(收敛是
107,因此根据您的系统/硬件,您的结果可能与此处给出的结果不同)。
而已。
有关单点计算的详细信息,请参见第6章。
本文地址:http://www.sd124.com/article/2018/1030/226642.html
《TURBOMOLE2016怎么安装 TURBOMOLE2016详细图文安装学习和补丁激活教程》由闪电下载吧整理并发布,欢迎转载!