
安装破解教程
1、在本站下载并解压,得到moe2015_10_installer_win.exe安装程序和crack破解文件夹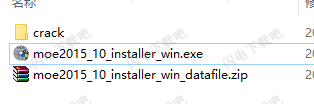
2、双击moe2015_10_installer_win.exe运行安装,如图所示,需要几分钟时间,耐心等待一会儿

3、进入安装向导,点击next
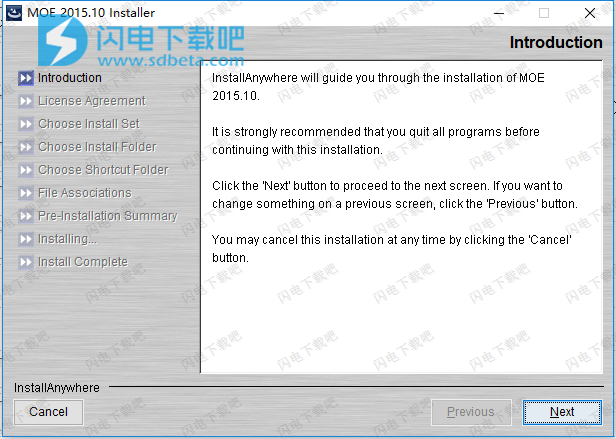
4、勾选我接受许可协议条款,点击next
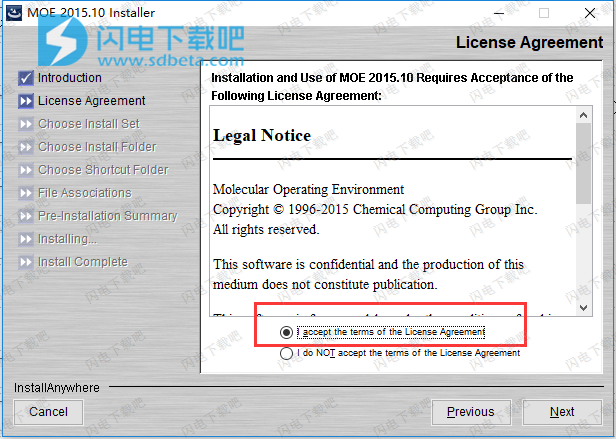
5、选择安装类型
全部安装
所有支持的操作系统的MOE可执行文件
(Windows.Linux.MacOS X)。 教育部手册。教程。 和示例文件。
单平台(Windows)
为Windos,MOE手册,教程和示例文件安装MOE可执行文件。我们选择第二项,点击next
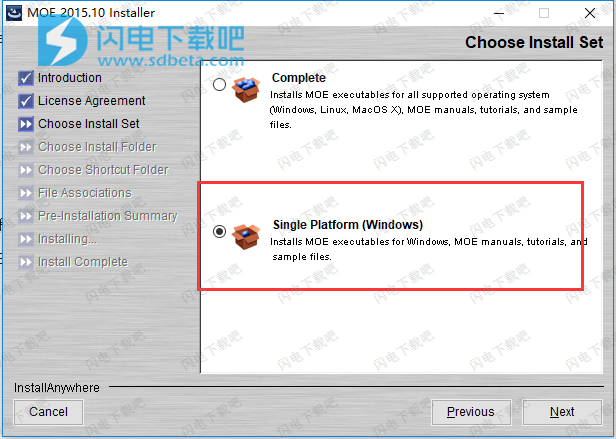
6、选择软件安装位置,点击next
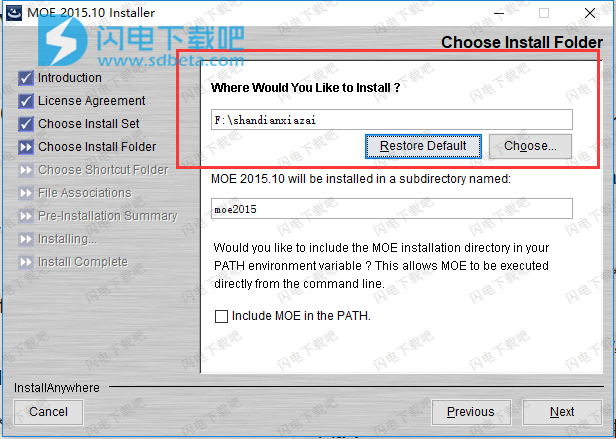
7、确认安装信息,并点击install安装
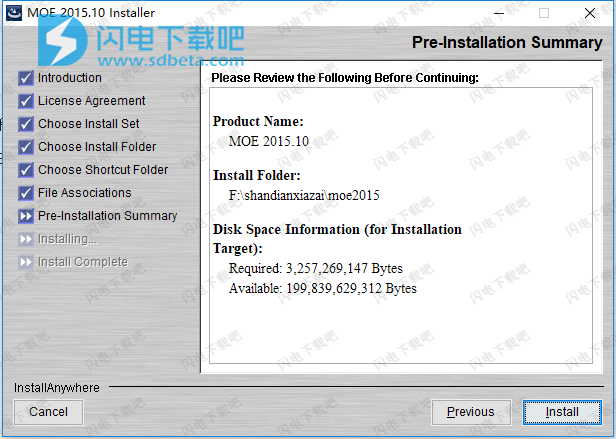
8、如图所示,软件安装中,这个过程也是需要一段时间
9、安装完成,点击next
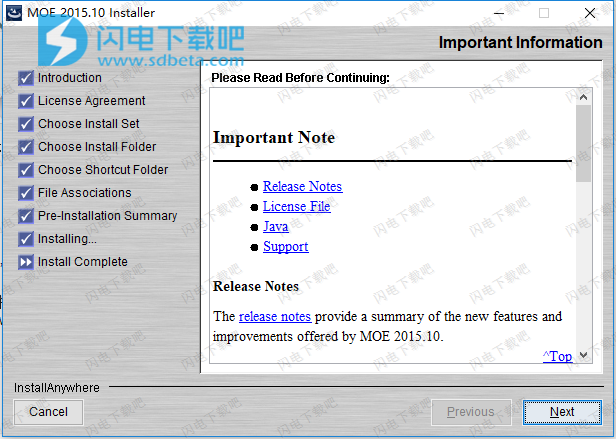
10、点击done退出向导
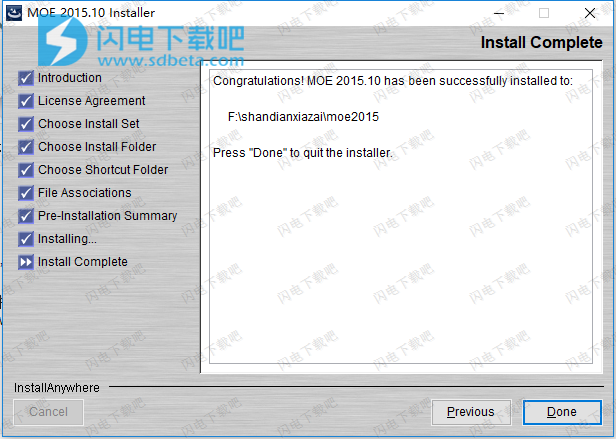
11、将crack_alternative文件夹中的patcher-win.zip进行解压得到patcher.exe,并将它复制到软件安装目录中,以以管理员身份运行,如图所示,点击patch按钮
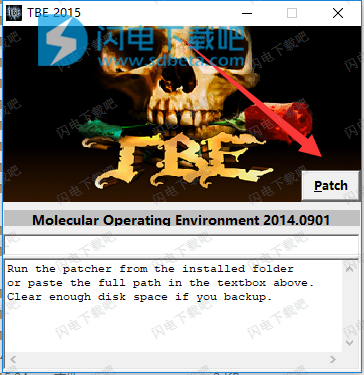
12、将license.dat文件复制到安装目录中,点击替换 c:\ moe2015 \ license.dat
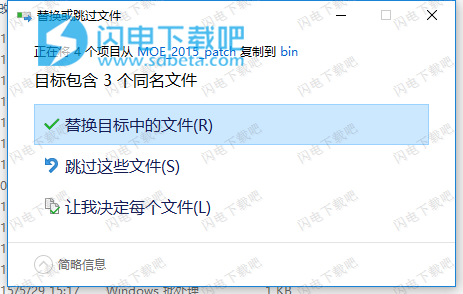
13、然后将crack中的MOE_2015_patch文件夹复制到软件安装目录,点击替换
功能特色
基于结构的设计
当可获得时,大分子晶体学数据可以是用于发现活性配体的有价值的信息来源。 MOE提供了一系列应用,用于可视化和理解受体活性位点和受体 - 配体相互作用的细节。 这些应用用于建议对候选结合物的配体或筛选配体数据库的改进。
1、主动站点检测
使用快速α形状算法检测和评分候选蛋白质配体和蛋白质 - 蛋白质结合位点。 这些网站的评分为配体结合倾向[Soga 2007]。 可视化各个站点或用“虚拟原子”填充它们,用于对接计算或从头配体设计工作的起点。
2、蛋白质:配体相互作用图
MOE - 基于结构的设计以图表形式显示与配体或一系列配体紧密接触的残留物[Clark 2007]。识别氢键,盐桥,疏水相互作用,阳离子-π,硫-LP,卤素键和溶剂暴露。浏览化学系列或受体家族系列,以确定选择性分析的保守或非保守相互作用。
3、联系统计,静电和交互图
构建由属性着色的分子表面。使用基于统计知识的电位预测接触偏好,并使用非线性Poisson-Boltzmann方程计算静电图。确定配体修饰的空间限制,热点和区域,并了解受体识别偏好。
使用3D-RISM进行溶剂分析使用3D-RISM [Kovalenko]计算水密度和结合去溶剂化惩罚图,这是基于液体密度泛函理论的溶剂化的第一原理理论。检测由相关性和空化效应产生的结合位点的非明显疏水区域,以优先化配体修饰。该方法比基于分子动力学或蒙特卡罗的替代方法快几个数量级。
4、脚手架更换
在受体活性位点的背景下生长R-基团,连接片段,转化和替换配体支架。使用基于反应的生物电子等排体转换转化结合的分子。杂交多个分子以产生新的候选结合物。搜索标准3D数据库或特殊支架和连接器数据库,以找到保留取代基几何形状的新型化学支架。添加额外的药效团特征以保持已知的支架相互作用或体积约束以满足形状要求。使用sdfrag应用程序生成自定义链接器数据库,或使用从CSD派生的链接器数据库(由CCDC分发)。
5、配体:接收器对接
将小分子停靠在大分子结合位点。提供构象数据库或动态生成构象。在各种评分函数[Corbeil 2012]之间进行选择,并可选择约束生成的姿势以满足药效团查询,从而将搜索偏向于已知的重要交互。使用简化的基于场景的界面来对接共价配体,运行电子密度引导对接或基于知识的模板引导对接。使用基于力场的方法使用MM / GBVI [Labute 2008]评分或基于快速网格的方法来细化姿势。提供了第三方对接程序的接口,用于高吞吐量虚拟筛选。使用MOE / smp技术并行化对接架构。
6、多片段搜索
多片段搜索是一种基于集合的方法,用于绘制受体结构中特定化学基团的优选位置[Miranker 1991]。大分子结构的活性位点填充有大量化学片段,其经受能量最小化方案。生成的组位置被聚类,评分(包括溶剂化效应)并写入数据库以供后续可视化和分析。
7、自动化结构准备
使用结构准备应用程序自动纠正晶体学数据中遇到的许多问题,例如缺失环,空残留物,链末端或断裂,缺少二硫键或原子名称,挑选替代构象等。通过导航不同的互变异构体/原体状态或使用Protonate3D [Labute 2008]自动优化氢键网络。 Protonate3D使用大规模组合搜索计算最佳质子化状态,包括滴定,旋转异构体和“翻转”。
8、用于配体优化的简化接口
CCG(与大型制药公司合作)开发了一个简化的界面,用于主动站点可视化和配体优化。 按钮栏提供结构准备,活性位点分析,分子特性/结合亲和力计算,潜在R组方向和活性位点配体优化的应用。 使用3D构建器或使用2D草绘器修改配体。 可视化和修改对齐的复合物,打开/关闭蛋白质,浏览对接姿势,药效团命中并使用系统管理器调整渲染。
基于片段的设计
MOE具有基于片段和从头设计应用的统一框架,可应用于基于配体和结构的设计项目。 MOE的药效团应用被整合到基于片段的设计方案中,该方案提供了一种有效的方法,用于在保持关键相互作用的同时产生新结构。
生长,连接,转化和替换配体支架(有或没有受体)
优化(灵活)活动站点中的坐标并计算绑定分数
应用MOE药效团过滤器以保持相互作用或官能团
自动应用2D和3D描述符,QSAR和指纹约束
直接搜索MOE .mdb或Omega .oeb文件并创建内部片段数据库
1、脚手架更换
MOE - 基于片段的设计通过结合创新的线性,环状或融合支架排列,替代支架[Grimshaw 2010]用于快速后续化合物。选择多个退出向量并执行同时搜索以生成面向多样性的支架。交换环键以识别和合并融合环排列。指定可选的连接向量以增加支架布置的范围并允许在多个连接点处进行环化。
2、配体杂交
将结合配体的片段(晶体结构或对接姿势)组合在受体活性位点的背景下产生新结构[Pierce 2004]。沿着所有重叠的“交叉”键,在所有配体对之间交换配体片段。可以限制交叉键以使产生的结构偏向合成可接近性。
3、片段链接和增长
MOE - 基于片段的设计在活动位点的上下文中的一个或多个位置生长配体或详细说明关键片段。链接片段以连接两个或多个独立片段。指定可选或基本连接点以提高灵活性。
4、药物化学转化
通过应用使用标准2D草绘器生成的反应转换规则(.rxn),对分子进行小的等量变化。 CCG提供了170多种药物化学转化的数据库。可以轻松地将自定义转换添加到数据库中,并且可以自动应用多个迭代来构建组合组合或在多代中进化转换以产生更复杂的片段和生物电子等排体。
5、自动过滤和评分
使用MOE的数百个描述符中的任何一个过滤生成的结构。应用SMARTS模式匹配以包括或排除分子中存在的子结构。应用QSAR,指纹或药效团模型作为额外的过滤器。使用基于形状的药效团查询基于配体的设计项目。使用MOE rsynth描述符估计“合成可行性”。细化(柔性)口袋中的结构并应用评分函数来估计结合亲和力。
6、片段数据库
使用MOE的SD管道工具创建内部片段数据库。枚举质子化状态并过滤输入SD文件以获得用于碎片的良好起始结构。仅保留具有铅样,非反应性等结构。使用算法组合碎片分子并生成构象。使用商业或定制的内部试剂作为反应引擎的输入以产生R-基团或支架库。
药效团发现
MOE包含业界领先的药效团发现应用程序套件,用于基于碎片,配体和结构的设计项目。药效团建模是生成和使用3D信息以搜索新型活性化合物的有力手段,特别是在没有可用的受体几何形状时。药效团方法使用广义分子识别表示和几何约束来绕过2D方法的结构或化学类偏差。
1、基于配体和结构的查询编辑器
使用易于使用的交互式查询编辑器使用蛋白质或配体构建查询。使用该查询筛选构象数据库以确定满足药效团模型的候选活性化合物。使用SMARTS化学模式和布尔表达式自定义药效团注释。限制具有体积约束的形状(受体或配体)并使用方向向量约束,原子质心或注释上的部分匹配来细化查询。应用MOE的活性位点分析工具有助于识别分子识别的关键相互作用。
2、自动药效团生成
通过考虑所有可能的离散几何和特征查询表达式的所有可能组合,从一组输入化合物(可能具有活动数据)查询和诱导分子排列。强制限制功能计数并添加自定义查询表达式。基于已知的活性化合物覆盖率,统计活动丰富度和匹配构象的原子重叠来对查询进行评分。蛋白质配体相互作用指纹(PLIF)可以在给定一组对齐的复合物(或对接结果)的情况下自动生成来自结合配体的查询。
3、构象数据库使用
MOE的并行化高吞吐量构象搜索应用程序构建用于从SD文件或SMILES字符串进行虚拟筛选的3D构象数据库。使用sdtools准备化合物集合,用于重复去除,互变异构体和电离状态以及立体枚举,片段生成和属性过滤。 CCG提供了一个大型的铅状全分子3D数据库和一个源自供应商目录和药物化学文献的大型片段数据库。
4、虚拟筛选
MOE - 虚拟筛选快速筛选构象数据库,以获得满足药效团查询的复合构象。搜索多个数据库,子范围的分子或对接化合物的数据库。直接搜索MOE MDB或Omega OEB文件。输出数据由满足3D药效团查询的分子组成(所有构象或仅满足查询的构象)。支持部分匹配,SMARTS模式,所有对称匹配的输出和基本特征的规范。在计算群集上以分钟为单位筛选公司集合。
5、基于配体和结构的脚手架更换
生长,连接和替换[Grimshaw 2010]配体支架(有或没有受体)。药物化学转化对依赖于使用标准草图产生的反应转化(.rxn)的分子进行小的等量变化。 CCG提供了170多个转换的数据库。如果可用,优化活性位点中的坐标(柔性受体)并计算结合分数,同时保留药效团特征。应用2D和3D描述符过滤器,QSAR和指纹模型。
药物化学应用
CCG在为铅生成和优化领域的药物化学家创建和部署解决方案方面拥有十多年的经验。分子操作环境(MOE)已被许多顶级药物研究公司采用,用于大规模药物化学部署。许多中小型制药公司也将MOE作为其主要的药物化学建模平台,以加速新疗法的开发工作。
适用于药物化学家和计算科学家的协调平台
多个不同发现项目组之间的无缝通信
易于与内部数据库,服务器和管道工作流系统集成
CCG(与大型制药公司合作)开发了一个简化的界面,用于主动站点可视化和配体优化。按钮栏提供结构准备,活性位点分析,分子特性/结合亲和力计算,潜在R组方向(替代机会)和活性位点配体优化的应用。使用3D构建器或使用2D草绘器修改配体。测量距离,角度和二面角轮廓。可视化和修改对齐的复合物,打开/关闭蛋白质,浏览对接姿势,药效团命中并使用系统管理器调整渲染
1、MOEsaic:用于配体分析的Web应用程序
MOEsaic是一种基于浏览器的应用程序,用于分析药物发现项目中的一系列小分子化学结构和相关属性数据。对齐分子以促进成对比较。进行子结构和相似性搜索。进行匹配分子对(MMP)分析。使用内置化学草图定义具有确定支架的R-基团。检测活动悬崖和生物电子等排。通过属性图和应用的过滤器可视化数据。使用文本和图像设计虚拟结构和文档结果。
2、可视化和分析非键合相互作用
使用扩展Hückel理论(EHT)可视化和分析配体 - 受体相互作用,如氢键,包括CH..O相互作用,卤素键,硫 - 氧相互作用,质子 - 和阳离子 - π相互作用。 EHT更准确地计算相互作用强度并考虑电子撤回和共振效应。
3、蛋白质 - 配体相互作用图
MOE - 药物化学应用自动生成与配体或一系列配体相互作用的活性位点残基的2D图[Clark 2007]。在2D中可视化关键的相互作用,如氢键,盐桥,疏水相互作用,阳离子-π,硫-LP和卤素键。使用描绘的空间轮廓识别配体取代的潜在位置。可视化溶剂暴露的配体原子和具有强疏水相互作用的残基。浏览化学系列或受体家族系列,以确定选择性分析的保守或非保守相互作用。
4、曲面和地图
构建由属性着色的分子表面,以定义和表征活性位点拓扑并识别配体取代机会。预测基于知识的非键合接触偏好或使用非线性Poisson-Boltzmann方程计算静电图以识别高值疏水区域和极性热点。使用3D-RISM计算水密度和结合去溶剂化惩罚图,3D-RISM是基于液体密度泛函理论的溶剂化的第一原理理论。检测由相关性和空化效应产生的结合位点的非明显疏水区域,以优先化配体修饰。
5、构象搜索和分析
探索配体构象空间,以获得有关生物活性构象和分子内相互作用的见解。使用LowModeMD [Labute 2010]通过执行快速隐式振动分析和短分子动力学模拟来生成大环和多组分系统的构象(例如,显式水或反离子)。
6、多分子的灵活比对
执行已知和推定配体的3D比对(或叠加)以确定生物活性的结构要求 - 特别适用于基于配体的药物设计方案,因为对齐的基团可能对于确定生物活性构象是重要的。使用全原子灵活对齐程序[Labute 2001],该程序结合了力场和基于形状和药效团特征的高斯描述的3D相似性函数,以产生小分子集合的可能对齐的集合。
8、脚手架更换,成长和片段链接
生长配体,连接片段并替换支架[Grimshaw 2010],用于快速后续化合物,包括创新的线性,环状或融合支架排列。在(柔性)活性位点中细化新结构,同时保持重要的药效团相互作用并计算预测的结合亲和力。使用药物化学变换通过对配体进行小的等量变化来探索局部SAR。 CCG使用从化学文献中提取的规则提供170多个功能组,同源和异构转换的数据库。可以使用标准2D草绘器添加新的转换。
9、Pharmacophore Discovery MOE - 药物化学应用
MOE包含业界领先的药效团发现应用套件,用于基于片段,配体和结构的设计项目。药效团建模是生成和使用3D信息以搜索新型活性化合物的有力手段,特别是在没有可用的受体几何形状时。药效团方法使用广义分子识别表示和几何约束来绕过2D方法的结构或化学类偏差。
使用交互式编辑器从分子比对或受体结构构建3D查询。执行构象数据库的虚拟屏幕以确定候选活性化合物。使用SMARTS化学模式(针对特定组)和/或表达定制药效团特征。通过使用包括,排除和外部体积的球体结合来限制形状(受体或配体)。使用原子上的方向向量约束或要素上的部分匹配来优化查询。
10、分子描述符计算
超过400种2D和3D分子描述符,包括拓扑指数,结构键,E-状态指数,物理性质,拓扑极性表面积(TPSA)和CCG的VSA描述符[Labute 2003],广泛适用于生物活性和ADME财产预测。应用基于扩展Hückel的描述符(例如LogP,LogD和摩尔折射率)来计算分子属性。计算小分子的pKa和pKb,并确定在给定pH下配体质子化状态的群体。使用描述符进行分类,聚类,过滤和预测模型构建。使用MOE的内置科学矢量语言添加自定义描述符。
11、Virtual Library Builder
通过基于反应的Combinatorial Library Builder枚举化合物库。使用商业或定制的内部试剂作为反应引擎的输入。进行简单的酯化反应或多组分Ugi型或Groebke-Blackburn-Bienyame反应。使用标准2D草绘器指定反应或多个同时反应步骤。自动筛选与靶标或药效团模型的化学相似性的反应产物。使用化学描述符或Lipinski的五级规则过滤结果以获得药物相似性。通过应用QSAR或药效团模型计算聚焦库。
本文地址:http://www.sd124.com/article/2018/0929/226008.html
《Molecular Operating Environment (MOE)20152015.10下载安装学习图文教程》由闪电下载吧整理并发布,欢迎转载!
相关电脑软件
- 相关文章:
- 相关软件:
本周热点
本月热点